| Trends重磅综述:以胰腺癌中的铁死亡作为靶标是把双刃剑 | 您所在的位置:网站首页 › hspa5 gene › Trends重磅综述:以胰腺癌中的铁死亡作为靶标是把双刃剑 |
Trends重磅综述:以胰腺癌中的铁死亡作为靶标是把双刃剑
|
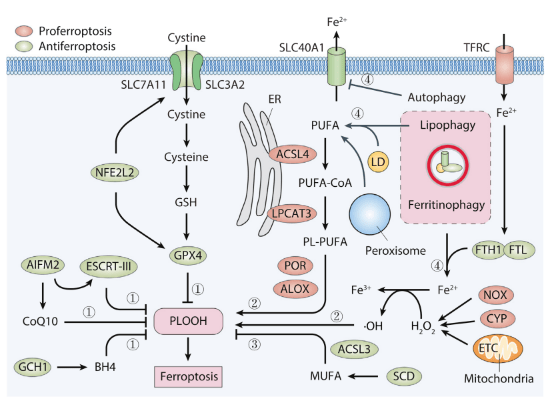
活性氧ROS的产生 活性氧ROS可以作为单一的分子去激活包括铁死亡在内的多种细胞死亡形式。由超氧化物驱动、铁离子催化的芬顿反应(Fenton reaction)是铁死亡中ROS产生的主要来源。由于细胞内的自由铁水平受铁摄入、储存、使用、分配、运出等动态调控,对于细胞内铁积累的任何扰动都有可能影响自由铁的水平以及随后ROS的产生,因此最终影响铁死亡的敏感性。例如,运铁蛋白受体(TFRC)过度表达带来的铁摄入增加会促进铁死亡。而对于组织内铁稳态的调控或许会进一步增加活体内铁死亡应答的复杂性。其他来源的ROS对于铁死亡的诱导作用包括但不仅限于线粒体的电子传递链、NADPH氧化酶(NOX,包括NOX1、CYBB/NOX2、NOX4),以及细胞色素P450酶(CYP)。由此带来更进一步的挑战就是难以区别不同来源的ROS始由对铁死亡有着同样的诱导效率。 然而,我们对于确定细胞命运的氧化压力精确阈值仍然一片未知。 脂质超氧化反应 由ROS引起的脂质超氧化反应当前被认为是铁死亡的生物化学特征。细胞膜或细胞器膜尤其容易受到脂质超氧化反应的影响,因为它们都有着较多的多不饱和脂肪酸(PUFA)。铁死亡PUFA衍生物的产生需要两种内质网中的酶:酰基辅酶A合成酶长链家族成员4(ACSL4)和溶血磷脂酰胆碱酰基转移酶3。溶血磷脂酰胆碱酰基转移酶3可使由脂肪氧合酶(ALOX)家族介导的脂质超氧化反应产生有毒的磷脂氢过氧化物(PLOOH)。相反,酰基辅酶A合成酶长链家族成员3(ACSL3)或者硬脂酰CoA去饱和酶(SCD/SCD1)介导的单不饱和脂肪酸(MUFA,如十八烯酸)的产生和激活可竞争性地抑制PUFA相关的铁死亡。 这体现出了铁死亡中不同脂质功能上的异质性。 除了内质网,过氧化物酶体中过缩醛磷脂的生物合成和脂噬带来的脂滴降解提供了铁死亡所需的PUFA额外来源。ALOX 是一个非血红素含铁酶家族,以细胞类型依赖的方式促进铁死亡。细胞色素P450氧化还原酶(POR)在铁介导的芬顿反应场合中介导脂质超氧化反应,扮演着一个ALOX非依赖的角色。 因此,除了自由铁离子可以引发非酶促的ROS产生,多种酶也参与到了铁死亡的过程中。 而我们对脂质- 蛋白质相互作用以及成孔蛋白质在铁死亡过程中的可塑性的了解仍然有限。 抗氧化防御 虽然高温、缺氧等环境压力可引发铁死亡,但经典的铁死亡诱导剂(如erastin 和 RSL3)可以通过抑制系统 xc−、谷胱甘肽 (GSH) 、谷胱甘肽氧化酶 4(GPX4) 形成的抗氧化轴心起作用。系统xc-,作为氨基酸运载体,由溶质载体家族 7 成员 11 (SLC7A11) 和溶质载体家族 3 成员 2 (SLC3A2) 组成,将胱氨酸运进细胞。胱氨酸随后通过还原反应被迅速转化成半胱氨酸并且用作谷胱甘肽GSH的合成。GPX4需要GSH作为辅因子去减少有毒的PLOOH,使其成为细胞膜中无毒的磷脂酒精PLOH。而SLC7A11在铁死亡中的活性和表达受到各种结合伴侣的双重调节。 除了GSH,其他细胞内的抗氧化物,例如辅酶Q11(CoQ11)、四氢生物蝶呤(BH4)、褪黑色素,可以通过它们强力的自由基捕获抗氧化能力阻止铁死亡中脂质超氧化反应的发生。在分子层面,线粒体相关凋亡诱导因子2 (AIFM2/FSP1) [30,31] 和 GTP 环化水解酶 1 (GCH1)分别负责还原性分子CoQ10 和 BH4的产生。然而,与 GPX4 抑制剂不同,单独的 AIFM2 抑制剂不足以诱导癌细胞中的铁死亡,这说明还有其他的冗余机制防止铁死亡的发生。过度的脂质超氧化反应最终会导致膜破裂,而这可被运转所需的内体分选复合体III(ESCRT-III)机制修复。这一ESCRT-III由钙元素引发的,是裂解细胞死亡中常见的一种膜修复机制。在PANC1细胞中对于ESCRT-III复合物已知的关键成员的敲低实验,包括敲低带电多泡体蛋白5(CHMP5)和带电多泡体蛋白6(CHMP6),增强了erastin/RSL3诱导的铁死亡。在转录层面,NFE2L2/NRF2在促进抗铁死亡基因的表达中扮演着核心角色。这些抗铁死亡基因包括SLC7A11、GPX4和其他细胞保护基因。 PDAC中铁死亡的调控 铁死亡可从表观遗传、转录、转录后等多层面进行调控。
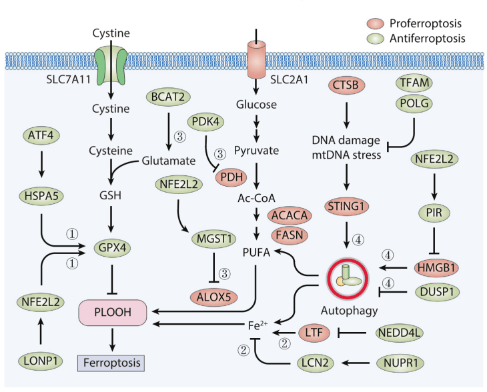
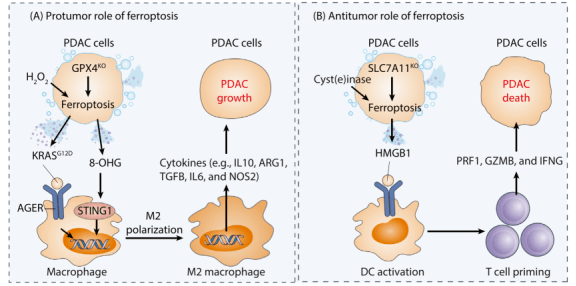
GPX4表达 在人类PDAC细胞中(PANC1、CFPAC1和MiaPaCa2),erastin通过激活性转录因子4(ATF4),一种内质网压力中介导基因表达的关键性转录因子,诱导热激蛋白家族A(HSP70)成员5(HSPA5)的表达。作为一种内质网相关的分子伴侣,HSPA5通过直接结合并防止GPX4蛋白降解从而抑制铁死亡。相反,遗传学(利用shRNA)或药理学(利用EGCG)对于HSPA5或ATF4的抑制会促进GPX4的降解,因此在PDAC离体细胞或异种移植小鼠模型上增强erastin诱导的铁死亡性细胞死亡。 青蒿琥酯(artesunate),一种获准用于初步治疗严重疟疾的药物,通过诱导铁依赖性的氧化损伤表现出促进铁死亡性抗癌活性。敲低HSPA5同样会抑制青蒿琥酯介导的GPX4蛋白上调,因此回复PDAC细胞对体内/体外青蒿琥酯介导的铁死亡的敏感性。由于要么自噬要么泛素-蛋白酶体系统 (UPS) 途径会在铁死亡过程中介导 GPX4 蛋白降解,HSPA5 介导的 GPX4 蛋白稳定性的确切机理仍有待查明。一种假说认为,HSPA5会激活雷帕霉素(mTOR)激酶信号通路的靶标,从而抑制GPX4铁死亡过程中的自噬性降解。在转录层面,线粒体蛋白酶离子肽酶 1 (LONP1)抑制NFE2L2介导的GPX4基因表达,从而促进人类PDAC细胞中erastin诱导的铁死亡。这些发泄在分子层面建立了线粒体功能紊乱和GPX4基因表达之间的联系。 然而,现在仍不清除 LONP1如何调控 NFE2L2的稳定化和随后的激活。 ALOX5活性 在哺乳动物细胞中,ALXO家族的六位成员被认为选择性地参与到了铁死亡的调控中。例如,ALOX5、ALOXE3、ALXO15和ALXO15B在BJeLR、HT-1080或PANC1细胞中有利于铁死亡的发生;ALOX15和ALOX12介导H1299细胞中的TP53依赖性铁死亡。ALOX促进脂质超氧化反应的能力严格受控于它们的结合蛋白。例如,微粒体谷胱甘肽S-转移酶(MGST1)是一种膜结合蛋白,有着重要的氧化还原和解毒活性。MGST1可以选择性地结合ALOX5去限制CFPAC1和PANC2.03细胞中毒性脂类的产生。这些毒性脂类产生在erastin或RSL3治疗后。此外,以NRF2最为人熟知的NFE2L2可以在PDAC细胞中通过它的抗氧化应答元件直接上调MGST1的基因表达。在PDAC病人中,MGST1的表达时常跟不良预后有关。 这些发现说明,以 MGST1介导的 ALOX5抑制为靶标的手段可能会成为促进 PDAC细胞铁死亡的可行策略。我们期待特定的MGST1抑制剂在未来几年可被开发用作基于铁死亡的治疗方法。 自噬降解 自噬对于细胞死亡形式的影响取决于被降解的第五(蛋白或细胞器)是否具有促进生存或促进死亡的功能。铁死亡被广泛认定为一种自噬依赖性的细胞死亡通路,因为铁储存蛋白铁蛋白的自噬周转(即铁蛋白自噬)有利于小鼠胚胎成纤维细胞和 PANC1 细胞中的erastin诱导的铁死亡。此外,在 PANC1 细胞中,溶质载体家族 40 成员 1(SLC40A1/铁转运蛋白)的自噬降解增加了细胞内游离铁的积累,导致芬顿反应介导的 ROS的 产生,并驱动铁死亡。SLC40A1同样可以在巨噬细胞中通过UPS通路被降解,这说明一个完整的降解通路或许可影响铁死亡的敏感性。相反,NEDD4样E3泛素蛋白连接酶(NEDD4L)介导的蛋白酶体降解乳转铁蛋白 (LTF) 蛋白可减少 PANC1 细胞中的铁死亡,进一步强调了非自噬降解系统在铁死亡中的影响。 PDAC细胞中自噬依赖性铁死亡的活性可以通过ROS传感器、DNA传感器和RAS信号进行微调。例如,pirin(PIR)不仅是NFE2L2靶基因,而且还是铁依赖性核氧化还原传感器。RIP的上调促进了PDAC细胞中铁死亡的抗性,这依靠限制氧化性DNA损伤和随后从细胞核向细胞质释放的高迁移率族框 1(HMGB1)来实现。细胞质HMGB1通过与beclin 1(BECN1)结合促进自噬依赖性铁死亡。BECN1是 III 类磷脂酰肌醇 3-激酶 (PI3K-III)复合物的核心成分,在促进自噬体的形成中起重要作用。又或者,在 PDAC 细胞中,erastin 诱导的溶酶体蛋白酶组织蛋白酶 B (CTSB) 或zalcitabine诱导的线粒体压力在细胞核内的累积,会导致DNA损伤和下游STING1/STING依赖的自噬通路的激活,导致铁死亡。除了能介导炎症和免疫反应,STING1还能通过促进微管相关蛋白1轻链3(MAP1LC3/LC3)的脂化,作为一种自噬体形成的直接调节剂。这些研究表明,细胞质中核DNA或线粒体 DNA(mtDNA)的异位存在可以通过激活STING1通路启动自噬依赖性的铁死亡。由于STING1基因在各类人类肿瘤中存在突变,因此确定热点STING1突变对铁死亡的影响会是件有趣的事情。相比之下,作为一种磷酸酶和RAS信号传导的调节剂,双特异性磷酸酶 1 (DUSP1)的激活会在体外或异种移植小鼠模型中损害PANC1和MIAPaCa2细胞中erastin或RSL3诱导的自噬依赖性铁死亡。可以很合理地认为,DUSP1通过抑制Ser555处的Unc-51样自噬激活激酶1(ULK1)和Ser15处的BECN1的磷酸化来限制自噬体的形成。 这些上面提到的机制说明了多种信号通路如何与自噬依赖性铁死亡的调控相互作用。 代谢途径 在PDAC细胞中,代谢过程极大地影响药物反应。虽然通过溶质载体家族2 成员1(SLC2A1)摄取葡萄糖限制了细胞凋亡,但它在各种人类PDAC细胞系或原代细胞中有助于系统xc-抑制剂(而非GPX4抑制剂)诱导的铁死亡。这是因为系统xc-抑制剂,而不是GPX4抑制剂,选择性抑制丙酮酸脱氢酶激酶 4(PDK4)表达。PDK4通过磷酸化丙酮酸脱氢酶(PDH)来抑制线粒体中的丙酮酸氧化,从而阻断PDAC细胞(PANC1和MIAPaCa2)中的铁死亡。代谢测定已证实丙酮酸氧化在PANC1细胞中生成乙酰辅酶 A,用于随后由乙酰辅酶A羧化酶α(ACACA)和脂肪酸合酶(FASN)的脂肪酸合成。最后,脂肪酸的增加为氧化应激下ALOX5介导的脂质过氧化提供了更多的底物。与由丙酮酸氧化或谷氨酰胺分解介导的线粒体促铁凋亡作用相反,支链氨基酸转氨酶 2(BCAT2)介导的谷氨酸转化和随后的GSH合成抑制由系统性xc-抑制剂诱导的铁死亡。铁蛋白吞噬和随后的AMP活化蛋白激酶(AMPK)通路的激活抑制了胰腺癌AsPC-1细胞中BCAT2的表达,表明AMPK在铁死亡中起双重作用。 这一双重作用取决于肿瘤类型和药物刺激。 研究表明,胰腺癌细胞铁死亡期间,一些特殊的因子控制着细胞内自由铁的水平。具体来说,核蛋白1(NUPR1)是一种转录调节因子,它在各种胰腺癌细胞死亡中充当应激蛋白。NUPR1的诱导表达,反式激活了编码铁输出蛋白的lipocalin 2(LCN)基因,从而减少了erastin-或 RSL3- 在 PDAC 细胞中诱导的铁积累,以及随后的铁死亡细胞死亡。而NUPR1抑制剂,ZZW-115,可在PDAC异种移植模型中增强咪唑酮橡皮蛋白 (IKE) 的抗癌作用。临床前和临床试验均表明NUPR1和LCN2可能在 PDAC 中发挥致癌基因那样的作用。这使得该通路成为潜在的治疗靶点。此外,细胞表面粘附受体CD44不仅通过介导包括PDAC在内的各种癌症类型中的铁内吞作用来增加细胞内游离铁水平,还可以通过与SLC7A11相互作用来增强GSH合成。 这些发现都表明CD44 在调节铁死亡中具有双重作用。 胰腺肿瘤形成中的铁死亡 转基因模型小鼠被广泛用于 PDAC发病机理的探索。这些小鼠包括Pdx1-Cre;KrasG12D/+小鼠(称为“KC小鼠”)和 Pdx1-Cre;KrasG12D/+;Tp53R172H/+小鼠(称为“KPC小鼠”)。虽然GPX4和SLC7A11是关键的铁死亡抑制因子,但KC或KPC中胰腺Gpx4或Slc7a11的条件性删除会在胰腺肿瘤发生过程中产生相反的表型。就像给小鼠喂食高铁饮食一样,胰腺中的Gpx4的有条件删除会加速cerulean或 L-精氨酸诱导的胰腺炎、KRASG12D驱动的小鼠胰腺肿瘤发生,而这些表型通可以过铁死亡抑制剂liproxstatin-1的治疗逆转。8-羟基-2'-脱氧鸟苷是铁死亡过程中DNA氧化损伤的主要产物。值得注意的是,这种8-羟基-2'-脱氧鸟苷会通过 STING1 介导的免疫和炎症反应来增加PDAC中的巨噬细胞浸润。相比之下,在KC小鼠中,通过脂质体氯膦酸盐治疗或通过改变Sting1−/−来抑制STING1信号以此消耗巨噬细胞,可防止Gpx4消耗或高铁饮食加速肿瘤发生。铁死亡性PDAC细胞可以通过自噬介导的外泌体分泌释放KRASG12D蛋白,从而导致巨噬细胞极化为促进肿瘤生成的M2表型。这种作用由细胞外KRASG12D蛋白对糖基化终产物特异性受体(AGER,也称为 RAGE)产生作用来介导。GPX4 的高表达与 PDAC 患者的总生存期呈正相关。这些发现重新强化了这样一种观点:由细胞死亡(例如,坏死性凋亡和细胞焦亡)诱导的炎症相关免疫抑制,可以加速 KRAS 驱动的胰腺肿瘤发生。
相反,胰腺 Slc7a11 的删除或者施加胱氨酸酶,可以限制胱氨酸摄取环节下游的CoA和 GSH合成,从而抑制KPC小鼠的肿瘤发生。目前尚不清楚在删除Slc7a11后KPC小鼠中哪些肿瘤免疫细胞或信号发生了变化。与GPX4不同,SLC7A11对PDAC患者没有预后作用。不管SLC7A11在氨基酸代谢中的多重作用如何,TP53 突变可能是肿瘤形成与抑制之间的一个检查点。 这种肿瘤形成由铁死亡介导,但其抑制机理目前仍未知。先前的研究表明TP53 的状态决定了自噬在胰腺肿瘤发展中的双重作用,而TP53 依赖性自噬是否调节、以及如何调节铁死亡在 PDAC 发病机制中的作用,这一点仍有待阐明。 PDAC治疗中的铁死亡 许多药物可以通过靶向调节机制在PDAC细胞中诱导铁死亡。具体来说,重新利用两种临床使用的药物,抗疟药物青蒿琥酯(artesunate)和抗HIV 1型病毒药物扎西他滨(zalcitabine),作为铁死亡诱导剂,或许在PDAC治疗方面有着诱人的吸引力。除了增加 PDAC 细胞中溶酶体氧化还原活性铁的水平外,青蒿琥酯还能够在非PDAC细胞中触发内质网应激和铁蛋白吞噬,从而促进铁死亡。临床前研究表明,扎西他滨通过抑制mtDNA的复制和修复,以此来触发人类PDAC细胞中的mtDNA损伤和接下来STING1介导的铁死亡。这一过程有可能作用DNA聚合酶 γ、催化亚基(POLG)或转录因子A、线粒体(TFAM)。但由于青蒿琥酯和扎西他滨对免疫系统都有潜在影响,因此有必要仔细评估这些药物对肿瘤微环境的副作用。 尽管其临床疗效不佳,但吉西他滨(gemcitabine)仍然是PDAC各个阶段治疗手段的基石。多项研究表明,联合使用铁死亡诱导剂可以克服癌细胞对于吉西他滨耐药性。详细来说,导致吉西他滨耐药性的其中一种机制与PDAC癌细胞中GPX4的上调有关。因此,通过抑制GPX4活性(例如,通过 RSL3)或诱导GPX4的降解(例如,通过EGCG或雷帕霉素),可以在体外实验或异种移植的PDAC模型中恢复或增强吉西他滨的抗癌活性。此外,若激活由SCD、F-box和包含WD重复结构域7(FBW7)和核受体亚家族4 A组成员 1(NRA41) 形成的枢纽,则阻止了铁死亡和细胞凋亡,最终导致PANC1和SW1990细胞的吉西他滨耐药性。高水平的SCD还与PDAC患者的总体生存率低有关,这从理论上增加了使用SCD抑制剂治疗PDAC的可能性。由于PDK4抑制葡萄糖介导的铁死亡敏感性,因此评估PDK抑制剂(如二氯乙酸盐)与吉西他滨联合治疗糖尿病PDAC患者将是一件有趣的事情。 总结 PDAC仍然是基础和临床肿瘤学面临的主要挑战,它以KRAS突变、较晚的诊断期和有限的治疗选择为特点。对于晚期的PDAC,我们需要更多的治疗手段。铁死亡是一种由不稳定铁的积累和过度的脂质超氧化反应驱动的非凋亡细胞死亡形式。这种细胞死亡形式的失调在各种疾病的发病机制中起着关键作用。尽管几项重要的研究进展提高了我们对PDAC各个阶段铁死亡的理解,但这些知识尚未转化为新的治疗方法以及临床有效的系统治疗或靶向治疗。多年来,许多选择性铁死亡激活剂得以被开发,尤其是以GPX4为靶向的激活剂。然而,尽管这些激活剂在细胞培养或动物模型中获得振奋人心的临床前效果,但这些激活剂均未通过临床验证。为了走出当前困境,通过高通量筛选和旧药新用的方法寻找代谢稳定的铁死亡剂会是促进临床转化的其中一个战略方向。和其他靶向疗法一样,癌细胞对于铁死亡的先天性的或获得性的抗药性已经成为一项重大挑战。因此,进一步阐明铁死亡的生物学基础、它与其他细胞死亡方式的动态关系、以及对铁死亡损伤反应的生物标志物,可能会找到PDAC临床管理的新方法。重要的一点是,我们在实现PDAC中刺激铁死亡的同时,应尽力避免致癌性炎症,诱导有效的抗癌免疫反应。目前,免疫检查点抑制剂已成为胃肠道中大多数其他实体癌的治疗基础。如果新型铁死亡诱导方案与免疫检查点抑制剂联合使用,有效的抗癌免疫反应会变得非常重要。虽然科学家已经提出了铁死亡的几个特征和生物标志物,但如何特异性和准确地量化铁死亡反应,尤其是在体内的铁死亡反应,仍然是一个重大挑战。 文章标题 : Targeting ferroptosis in pancreatic cancer: a double-edged sword (1) 以胰腺癌中的铁死亡作为靶标:一把双刃剑 DOI: https://doi.org/10.1016/j.trecan.2021.04.005 文献来源: 1. Chen X, Kang R, Kroemer G, Tang D. Targeting ferroptosis in pancreatic cancer: a double-edged sword. Trends Cancer. 2021 May 19; 2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018 Nov;68(6):394–424. 3. Huang J, Lok V, Ngai CH, Zhang L, Yuan J, Lao XQ, et al. Worldwide Burden of, Risk Factors for, and Trends in Pancreatic Cancer. Gastroenterology. 2021 Feb;160(3):744–754. 4. GBD 2017 Pancreatic Cancer Collaborators. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2019 Dec;4(12):934–947. 5. Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013 Jun;144(6):1252–1261. 6. 胡骐冰. 妇产科研究所 新闻动态 [Internet]. [cited 2021 Jun 23]. Available from: http://www.gdklmod.com/fuchanke/contents/223/866.html 8. Kroemer Lab. Home [Internet]. [cited 2021 Jun 23]. Available from: http://www.kroemerlab.com/ 通讯作者介绍:
唐道林教授 (图片来源:UT Southwestern) 唐道林博士出生于 1976年 7月,目前是美国匹兹堡大学外科系研究助理教授,匹兹堡大学肿瘤研究所损伤模式分子中心基础部主任和学术带头人,国际上 hmgb1和细胞自噬研究的学术带头人之一,首次阐明了 hmgb1分子与细胞自噬的关系及其在肿瘤中的作用,其实验室首次阐明了 hmgb1与细胞自噬的关系及其在人类疾病例如肿瘤中的作用。近五年来,在国际权威期刊发表论文六十余篇(6)。
康睿教授 (图片来源:UT Southwestern) 康睿教授,本科毕业于白求恩医科大学,硕博连读毕业于中南大学湘雅医院,在美国匹兹堡大学开展博士后训练,历任湖南省儿童医院医生、匹兹堡大学助理教授和副教授、广州医科大学特聘教授。2018年加入UT Southwestern 外科部门(7)。
Guido Kroemer, M.D., Ph.D. PU-PH (图片来源:Kroemer Lab) Guido Kroemer 教授,现任巴黎笛卡尔大学医学院教授、法国医学研究委员会(INSERM)“代谢、癌症和免疫”研究团队主任、古斯塔夫大学代谢组学和细胞生物学平台主任Roussy 综合癌症中心、Cordeliers 研究中心副主任和法国巴黎Hôpital Européen George Pompidou 的医院从业人员。他还是瑞典斯德哥尔摩卡罗林斯卡学院的外籍兼职教授(8)。 注:此推文未经许可禁止转载! 【科研吐槽】那些毕业季的坑,你入了吗? 【科研吐槽】复旦医生教你选择科室平台时如何“避雷”?这四种科室,请谨慎入坑! 焦虑的硕士毕业生,千万别对你的第一份工作无所谓! 上海顶尖三甲医院或已进入究极内卷时代——一位交大转行医生的自白 Cell对话顶级科学家:围观下大佬们眼中肿瘤研究领域的三大关键难题如果您或者科室有科研上的困扰 扫码备注:科研合作返回搜狐,查看更多 |



【本文地址】